Introducción
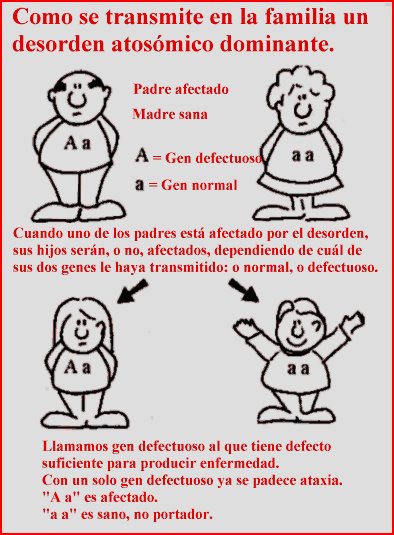
El síndrome de Blau es un trastorno inflamatorio genético autosómico dominante que afecta a la piel, los ojos y las articulaciones. Es causada por una mutación del gen NOD2 (CARD15).
 Los síntomas suelen comenzar antes de los 4 años y la enfermedad se manifiesta como sarcoidosis cutánea de inicio precoz, artritis granulomatosa y uveitis.
Los síntomas suelen comenzar antes de los 4 años y la enfermedad se manifiesta como sarcoidosis cutánea de inicio precoz, artritis granulomatosa y uveitis.
Historia
En 1985, Edward Blau, un pediatra en Marshfield, Wisconsin, informó que había una familia que durante cuatro generaciones había tenido inflamaciones granulomatosas de la piel, los ojos y las articulaciones. La enfermedad se transmitía como un rasgo dominante autosómico. El mismo año se documentó que una familia a lo largo de dos generaciones presentaba sinovitis granulomatosa, uveítis y neuropatías craneales. La condición se transmitió de manera autosómica dominante. En 1981 Malleson documentó que una familia tenía sinovitis autosómica dominante, camptodàctilament y iridociclitis. Una persona murió de artitas granulomatosa del corazón y de aorta. En 1982, Rotenstein informó de una familia con arteritis granulomatosa, erupción cutánea, iritis y artritis transmitidas como rasgo autosómico dominante durante tres generaciones. En 1990 Pastores documentó una relación con un fenotipo muy similar al que Blau había descrito y sugirió que la enfermedad se llamara síndrome de Blau (BS). También se mostraron las similitudes entre las familias antes mencionadas y la BS, así como las diferencias significativas en los fenotipos.
En 1996 Tromp realizó una búsqueda amplia al genoma con miembros afectados y no afectados de la familia original. Un marcador, D16S298, dio una puntuación LOD máxima de 3,75 y situó la susceptibilidad del Síndrome de Blau dentro del intervalo 16p12-Q21. Hugot se encontró un locus de susceptibilidad para la enfermedad de Crohn, una inflamación granulomatosa del intestino, el cromosoma 16 cercano al locus para Síndrome de Blau. A partir de la información anterior, Blau sugirió en 1998 que el defecto genético de la enfermedad pulmonar y de la enfermedad de Crohn podría ser el mismo o similar.
Finalmente en 2001 Miceli-Richard encontró el defecto del Síndrome de Blau en el dominio de unión a nucleótidos de CARD15 / NOD2. Comentaron en su artículo que también se habían encontrado mutaciones en el CARD15 en la enfermedad de Crohn. La confirmación del defecto que se encontraba en el Síndrome de Blau en el gen CARD15. en 2002 con la familia Síndrome de Blau y otros. Con esta información, el diagnóstico de Síndrome de Blau no sólo se determinó por el fenotipo sino por el genotipo.
La sarcoidosis de inicio precoz es Síndrome de Blau sin antecedentes familiares, el Síndrome de Blau ha sido diagnosticado en pacientes que no sólo tienen la tríada clásica sino granuloma en diversos órganos.
Causa
La elucidación que el defecto génico en el Síndrome de Blau implica en el gen CARD15 / NOD2 ha llevado a muchos investigadores, a definir cómo funciona este gen como parte del sistema inmunitario innato, que responde a los polisacáridos bacterianos, como el muramil dipeptide, para inducir vías de señalización que inducen respuestas de citocinas y protegen el organismo. En el Síndrome de Blau, el defecto genético parece conducir a la expresión excesiva, y un mal control de la respuesta inflamatoria provocando un granulomatoso, inflamación y daños a los tejidos.
Tratamiento
El tratamiento incluye los medicamentos antiinflamatorios habituales como los glucocorticoides suprarrenales, antitabolits y también agentes biológicos como anti-TNF y infliximab, todos con varios éxitos.

