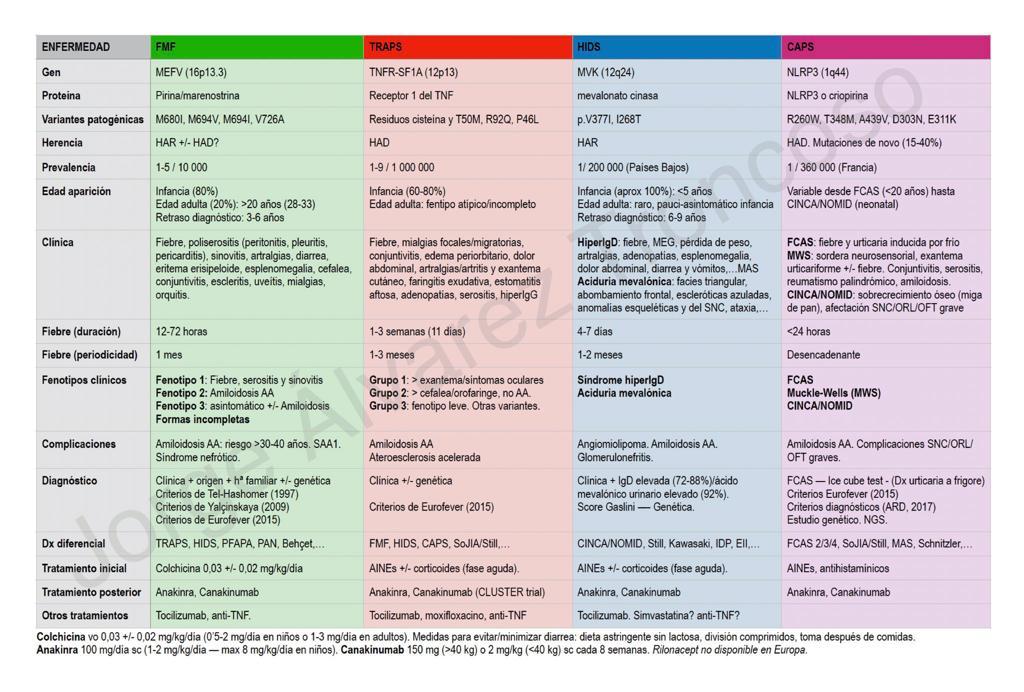
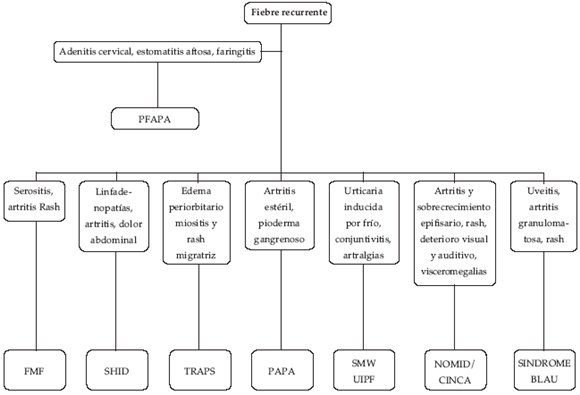
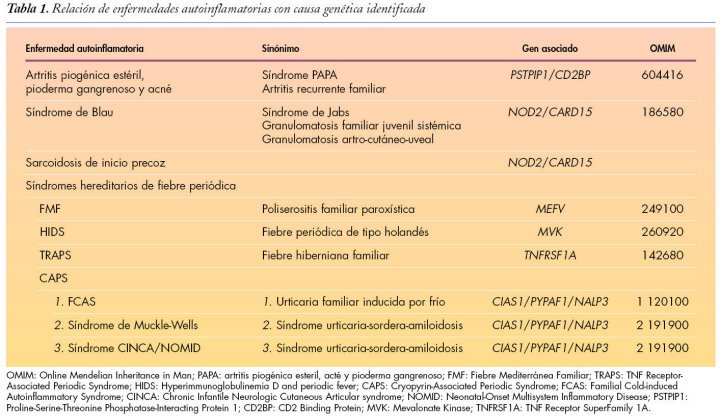
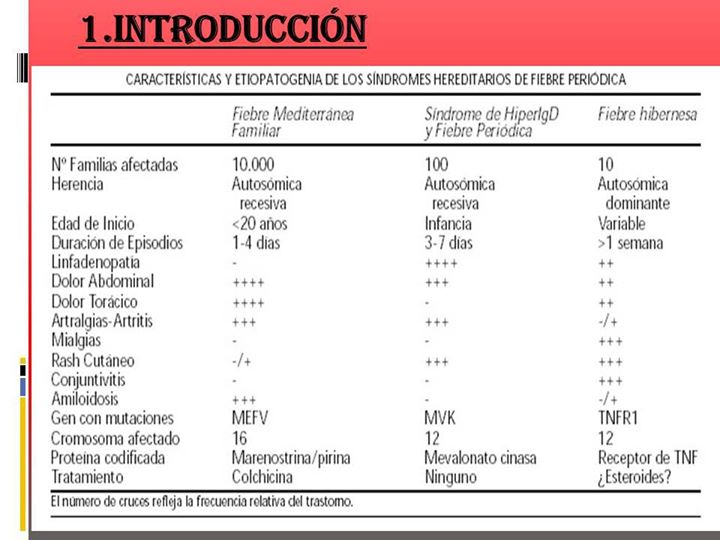
Consideramos la inflamación como un complejo proceso en el que intervienen una serie de células efectoras en reacción a estímulos físicos, químicos, biológicos e inmunológicos.
Sin embargo surge un nuevo concepto, el de los llamados “síndromes auto-inflamatorios”.
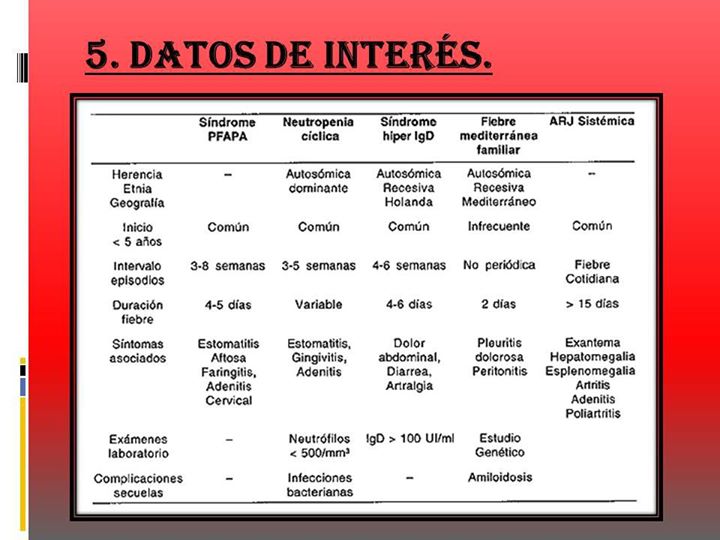
Dentro de estos síndromes se encuentran los denominados síndromes de fiebre periódica recurrente hereditarios:
Síndrome de fiebre periódica asociado al receptor del TNF
Síndrome crónico infantil neurológico, cutáneo y articular
Síndrome de Muckle Wells
Síndrome auto inflamatorio familiar por frio
Su característica fundamental es la recurrencia de la fiebre acompañada de algún otro síntoma de naturaleza inflamatoria como dolor abdominal, diarrea y otros síntomas.
Estos episodios son auto-limitados con una duración de unos días, reapareciendo recurrentemente, tras periodos libres de síntomas.
La importancia creciente de estos síndromes ha llevado a la creación de un registro internacional de mutaciones genéticas responsables de los mismos, a través de internet: Infevers.
Investigaciones llevadas a cabo recientemente han descubierto que algunas enfermedades febriles poco frecuentes son producidas por alteraciones genéticas.
En algunas puede haber varios familiares afectados.
¿Qué significa una anomalía genética?
Quiere decir que un gen ha sido modificado por un tipo de accidente llamado “mutación”. Esta mutación cambia la función del gen implicado, lo que produce una información errónea que origina la enfermedad. En las células de todos nosotros hay 2 copias de cada gen. Una copia se hereda de la madre y otra del padre.
¿Es el HIDS una enfermedad hereditaria?
Es un síndrome febril periódico hereditario.
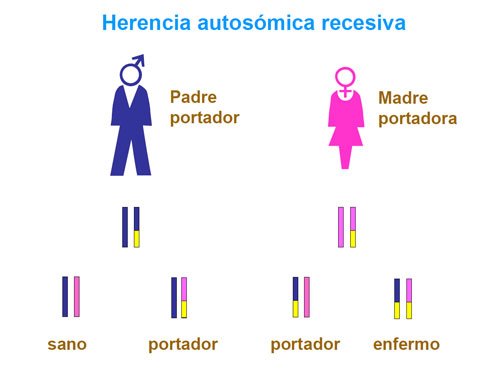
Como ocurre con casi todos los genes humanos, todas las células del organismo tienen 2 copias de MVK. Una copia es heredada de la madre, la otra del padre.
La fiebre periódica solo aparece cuando ambas copias de MVK están dañadas. Esto es conocido como herencia autosómica recesiva.
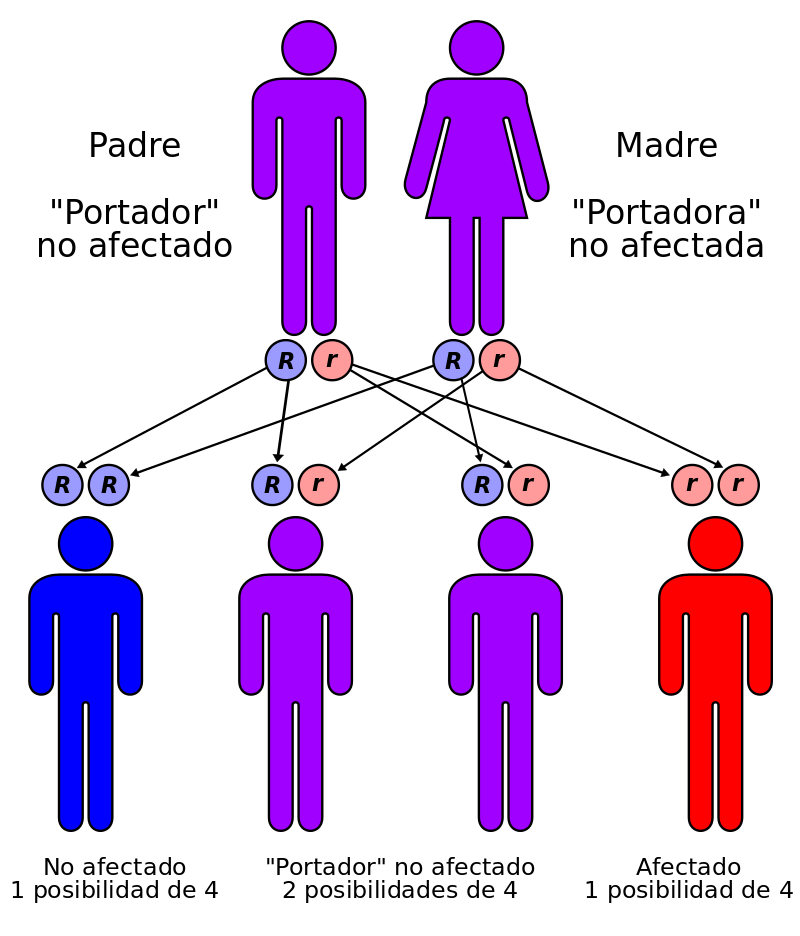
Tanto la madre como el padre tienen una copia defectuosa del gen MVK y, aunque ellos están sanos, pueden pasar los genes dañados a sus hijos.
Cada hijo de esta pareja tiene el 50% de posibilidades de ser un portador sano (transmitir la enfermedad sin padecerla) y un 25% de desarrollar la enfermedad.
Los hijos de los pacientes solo serán portadores sanos, a no ser que sus parejas también tengan una copia dañada del gen.
Las posibilidades de que ambos miembros de una pareja lleven un gen defectuoso aumentan cuando entre ellos existe relación de parentesco (consanguinidad)
En el caso del HIDS la herencia es recesiva.
Si la mutación está presente en los dos padres y es recesiva, significa que ambos padres llevan la mutación, pero solo uno de sus dos genes. Para que se produzca la enfermedad los dos genes tienen que estar afectados, por lo que ninguno de los padres estará enfermo. El riesgo de que el niño herede la mutación es del 25%.
De User:LordT modification from the original of en:User:Cburnett – Own work in Inkscape, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=3331122
Sinopsis del HIDS
La enfermedad periódica fue mencionada probablemente por primera vez en literatura médica 1806, cuando Heberden describió una entidad clínica en la cual el dolor periódico que afectaba el abdomen y a veces el pecho y las extremidades pronominadamente.
Estos pacientes sufrieron dolores que son regularmente intermitentes, que vuelven periódicamente, en intervalos cortos notables regulares.
El HIDS se nombró así en 1995, pero fue descrito en 1984 por Van der Meer y colaboradores Prieur y Griscell encontraron 8 pacientes con fiebres periódicas unidas con otras enfermedades, quienes más tarde presentaron un aumento de IgD en suero. En 1987, Marshall describió este síndrome con carácter agudo con aumento de IgD en niños con fiebre periódica, aftas, faringitis y adenopatías, que respondían a tratamiento con corticoides.
Inicialmente se describió el HIDS en pacientes de Holanda y otros países de Europa, más tarde en los EE.UU. Aunque la mayor parte de los casos descritos pertenecen a Europa Occidental, sobre todo a Francia y Holanda.
Clínicamente, el HIDS se semejaba a la Fiebre Mediterránea Familiar o el síndrome infantil articular cutáneo neurológico crónico. Este síndrome se caracterizó por ataques de fiebre recurrentes acompañados por el dolor abdominal, artralgia, dolor de cabeza y lesiones en la piel. Durante los episodios de fiebre, todos los pacientes tenían valores de IgD en el suero por encima de 60mg/l, medidos en 2 ocasiones con un mes de diferencia.
HIDS es parte de un grupo de los síndromes que tienen características en campo común, especialmente la fiebre recurrente. Estos se conocen colectivamente como “síndromes periódicos de la fiebre” o “síndromes autoinflammatory hereditarios”. El primer paso para su diagnóstico, es que el médico considere esta enfermedad rara.
HIDS es tan raro que muchos médicos saben poco o nada de esta enfermedad y no pensaran en este diagnóstico. La mayoría de los niños enfermos de HIDS, pueden contar una niñez con numerosos ingresos en el hospital y examinaciones medicas incontables. A estos niños se les pasara su infancia sin poder hacer cosas que hacen otros niños a su edad.
Una suspicacia de HIDS es consolidada por un curso típico de la enfermedad: ataques recurrentes de por vida de la fiebre sin una causa clara, en algunos días y que luego se resuelven espontáneamente, y los síntomas a veces similares en un hermano o una hermana. A veces, sin embargo el curso de la enfermedad es menos característico.
Debido a que los ataques que produce esta enfermedad ocurren con demasiada frecuencia, tiene un gran impacto en la vida social, laboral y cultural.
Los periodos recurrentes de la enfermedad afectan a la vida en familia.
En la mayoría de las familias, la vida se centra alrededor del paciente con HIDS que puede tener consecuencias desventajosas. Los pacientes con una enfermedad crónica tal como HIDS necesitan la ayuda, el reconocimiento y la información.
En ocasiones, el hablar con alguien que sabe lo que tienes es la mejor medicina.
Ocurre con hombres y mujeres con la misma frecuencia.
No causa muerte temprana.
Características del Síndrome de Hiperinmunoglobulinemia D
El Síndrome de Hiperinmunoglobulinemia D es una de las denominadas enfermedades raras y es un síndrome hereditario de tipo autonómico recesivo de fiebre periódica descrito por el Profesor Dr. Van der Meer en 1984. Se produce por al menos una mutación en el brazo largo del cromosoma 12q24 (gen MVK) lo cual lleva a producir un déficit parcial de una enzima llamada Mevalonato Quinasa.
Hasta el 2005 se han descrito unos 200 pacientes en todo el mundo.
Donde más casos se han descrito, ha sido en Europa Occidental, sobretodo en Francia y Holanda, aunque también en pacientes de otras razas.
Es una enfermedad muy rara que se halla presente al nacimiento.
Los síntomas suelen comenzar en la primera infancia, durante el primer año de vida.
En el 80% de los pacientes la enfermedad aparece en sus primeros 24 meses de vida.
¿Es la enfermedad igual en todos los pacientes?
Dependiendo de cuál sea la mutación la enfermedad puede ser leve o grave. Dentro de la misma familia la gravedad de la enfermedad puede diferir entre los distintos miembros afectados. Clínicamente se caracteriza por ataques recurrentes de fiebre alta de aparición brusca, náuseas, vómitos y dolor abdominal, presente en casi todos los pacientes; a la exploración los enfermos presentan palpación abdominal dolorosa.
Ya hemos dicho que se caracteriza por la aparición de recurrentes episodios de fiebre precedidos de escalofríos.
Generalmente, fuera de los episodios de la fiebre, una persona no se siente enferma. Algunos enfermos tienen un ataque una vez al mes, pero otros lo tienen más a menudo.
Sin embargo, otros pacientes lo tienen cada seis meses, pero la frecuencia de los ataques pueden cambiar en el curso de la vida. A veces, una persona no tiene ningún síntoma de enfermedad por meses y luego la enfermedad vuelve aparecer de nuevo. En mucha gente el patrón de la enfermedad cambia durante la pubertad o después de ella, y los ataques ocurren menos a menudo. Pero desafortunadamente esto no ocurre siempre.
Esta enfermedad no tiene una influencia directa en el crecimiento y desarrollo del niño, aunque los episodios frecuentes de la enfermedad pueden por supuesto tener una influencia negativa, por ejemplo con el tiempo que faltan a la escuela.
¿Cuál es la causa de la enfermedad?
La respuesta exacta a esta pregunta no se sabe todavía.
Sabemos que cierta proteína no funciona correctamente en una persona con HIDS debido a un defecto en la información hereditaria (esta proteína se llama Kinase del Mevalonate).
Es desconocida porqué esta proteína tiene que ver con la inflamación y la fiebre exactamente. Parece ser un defecto en el control de reacciones inflamatorias.
El cuerpo posee una inflamación complicada y un sistema inmune. Este sistema es tan complicado que los muchos de los detalles son inexplicados en esta fecha todavía.
Una de sus funciones es conseguir libre y rápidamente las sustancias que no pertenecen al cuerpo.
Por ejemplo, imagine una astilla en su dedo: si esto permanece en el lugar, dentro de un tiempo, una reacción inflamatoria comenzará en ese punto.
Usted notara esto porque ese punto se pondrá caliente y rojo, y dolerá.
Si se pone peor, usted puede incluso tener fiebre. Pero tan pronto como la astilla caiga hacia fuera, la inflamación no es necesaria y solamente es molesta.
El cuerpo por lo tanto tiene la capacidad de expulsar una reacción inflamatoria cuando no se necesita.
Para entender mejor como funciona esta enfermedad, he aquí un claro ejemplo; En una situación donde una persona sana, tiene facilidad para librarse de un virus que afecta a su sistema inflamatorio, en una persona que padece HIDS la inflamación conseguirá entrar y se quedará en su cuerpo, incluso después de que el virus haya desaparecido.
Por este motivo la mayoría de los médicos nos recomiendan paciencia y que se guarde reposo después de haber sufrido una crisis de la enfermedad.
Durante estas crisis se nota, una reacción inflamatoria severa con fiebre y por ejemplo dolor en los músculos y otros síntomas (no tiene porqué ser siempre los mismos en cada crisis, ni en cada paciente). Solamente después de un período largo, la inflamación se va y la persona se va encontrando cada día mejor, hasta el próximo ataque.
La razón exacta de los ataques en HIDS es confusa todavía.
Para acentuar: los pacientes con HIDS no tienen un sistema inmune deteriorado, pero si son más propensos que otros a coger bacterias o un virus.
A la hora de un ataque de la fiebre hay una reacción inflamación.
Los antibióticos por lo tanto no ayudarán a disolver todos los ataques de la fiebre. La exposición a las bacterias puede provocar probablemente un ataque de la fiebre.
Por ejemplo, muchos pacientes son propensos a tener un ataque de la fiebre cuando hay una gripe que circula: el virus de la gripe es probablemente el factor que acciona en este caso. El HIDS no es contagioso en ningún caso, ni por una persona con pocas defensas y tampoco en personas sanas.
Se desconoce como un defecto genético que está presente todo el tiempo origina una enfermedad que se manifiesta solo durante los ataques de fiebre.
Estos pueden aparecer espontáneamente o ser provocados por traumatismos leves, intervenciones quirúrgicas, infecciones sin importancia, estrés, tensión emocional o una simple vacuna. Por ejemplo, a menudo habrá un ataque después de un periodo de exámenes o incluso algo que afecte en la vida personal y sentimental.
Las mujeres con esta enfermedad, pueden tener ataques desencadenados por el ciclo menstrual. Cuando se quedan embarazadas, tienden a tener menos síntomas.
¿Cómo se diagnostica?
A pesar de las investigaciones existentes desconocemos como la falta de Mevalonato Quinasa produce fiebre e inflamación.
Durante los ataques hay una inflamación generalizada, como si tu cuerpo estuviera combatiendo una infección grave.
Esta inflamación produce fiebre, pérdida del apetito y malestar general, así como un aumento del aumento de leucocitos.
Dado que no hay una infección responsable de esta inflamación el HIDS se considera una enfermedad auto-inflamatoria.
Ya sabemos que es un error congénito del metabolismo de causa genética.
El gen afectado se llama MVK. Cada gen contiene instrucciones para la formación de una proteína específica; en el caso de MVK es la llamada Mevalonato Quinasa.
Esta proteína facilita una reacción química en el cuerpo, la transformación de un compuesto llamado Mevalonato y otro llamado FosfoMevalonato.
Este es uno de los pasos iniciales en la producción de un grupo importante de moléculas en nuestro cuerpo, entre las que se encuentra el colesterol.
Las enzimas genéticamente defectuosas afectan al metabolismo de nuestro cuerpo y causan enfermedades que se conocen como errores congénitos del metabolismo.
La gravedad de la enfermedad está relacionada con la severidad del déficit de la enzima.
Se establece en función de las características clínicas de la enfermedad junto con las pruebas bioquímicas o genéticas que definen el déficit de MVK.
Los niveles de IgD en suero están con frecuencia elevados aunque suelen ser normales en pacientes menores de 3 años.
El análisis genético revela una mutación en el gen MVK.
En una pareja con un hijo afectado las posibilidades de engendrar un nuevo hijo con la enfermedad son del 25% por lo que se recomienda el asesoramiento genético
La enfermedad se sospecha en base a sus manifestaciones clínicas.
El diagnostico se puede sospechar realizando una cromatografía (un tipo de análisis muy especial) en una muestra de orina recogida durante un ataque febril.
Los niños con la enfermedad tienen aumento de los niveles de ácido mevalonico.
En estos niños se puede medir la actividad de la mevalonato quinasa en células sanguíneas, aunque esta prueba solo está disponible en centros muy determinados.
El estudio genético puede hacerse con fines de investigación.
Dentro de los padecimientos de causa inmunológica, se encuentra el Síndrome de Hiper-IgD o Hiperinmunoglobulinemia D, escasamente conocido y diagnosticado dada su rareza y también su pobre gama e síntomas para su sospecha. Se le ha descrito dentro de los padecimientos que cursan con síndrome febril de tipo periódico.
Existe desafortunadamente, una limitante en cuanto a su diagnóstico definitivo, ya que dentro del laboratorio de Inmunología, no se tiene bien establecido el método de detección de los niveles de IgD, al menos en nuestro país.
La Inmunoglobulina D (IgD) es, de las proteínas presentes en el suero humano, la menos conocida en cuanto a su función biológica. En 1972, se descubrió la IgD.
¿Qué análisis o pruebas son útiles?
La analítica puede demostrar una elevación de los reactantes inflamatorios (VSG y PCR) durante los ataques. Los niveles de IgD (una inmunoglobulina circulante) en suero están con frecuencia elevados, aunque pueden ser normales en los primeros estadios de la enfermedad.
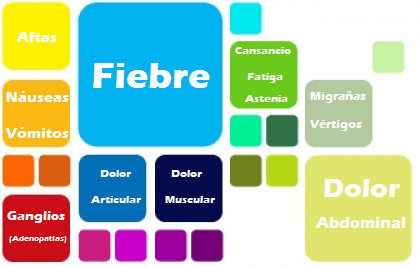
¿Cuáles son sus Síntomas?
“Un ataque o Brote” es anunciado por las frialdades, seguidas por una subida aguda de la temperatura del cuerpo y dura de cuatro a seis días, con desaparición gradual.
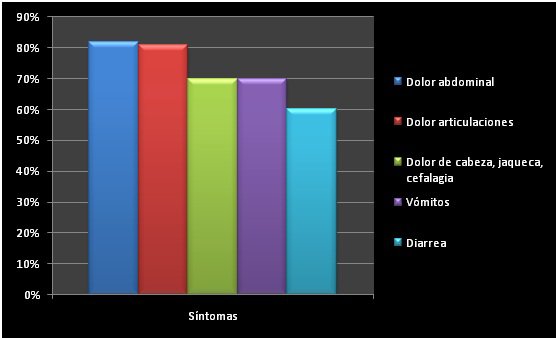
El dolor cervical y el abdominal, con vómitos, diarreas o ambas a la vez acompañan casi siempre el ataque.
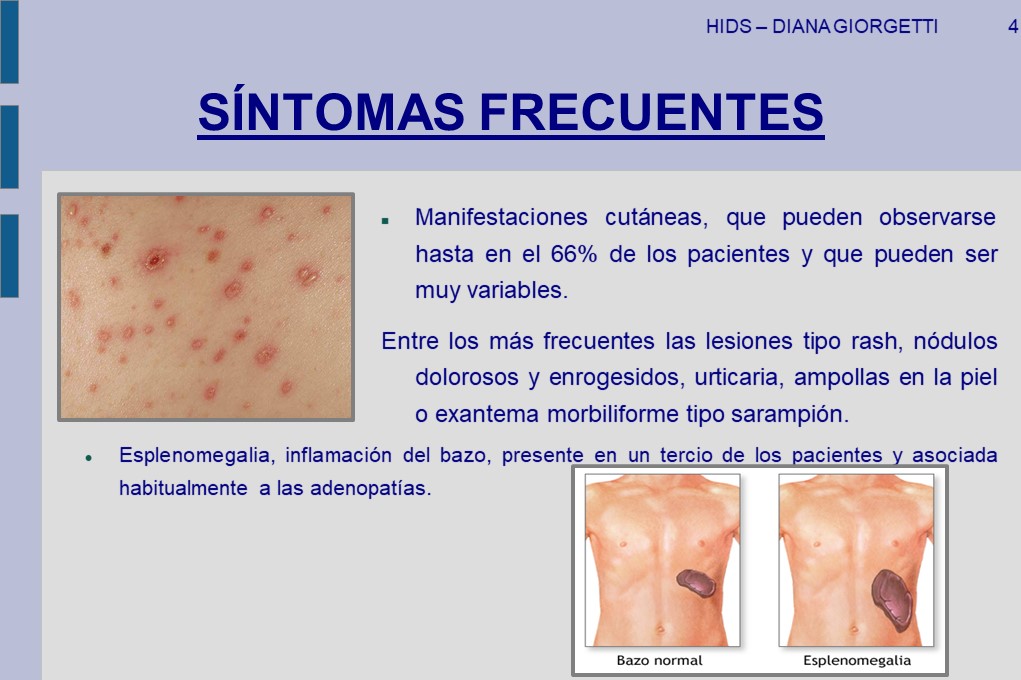
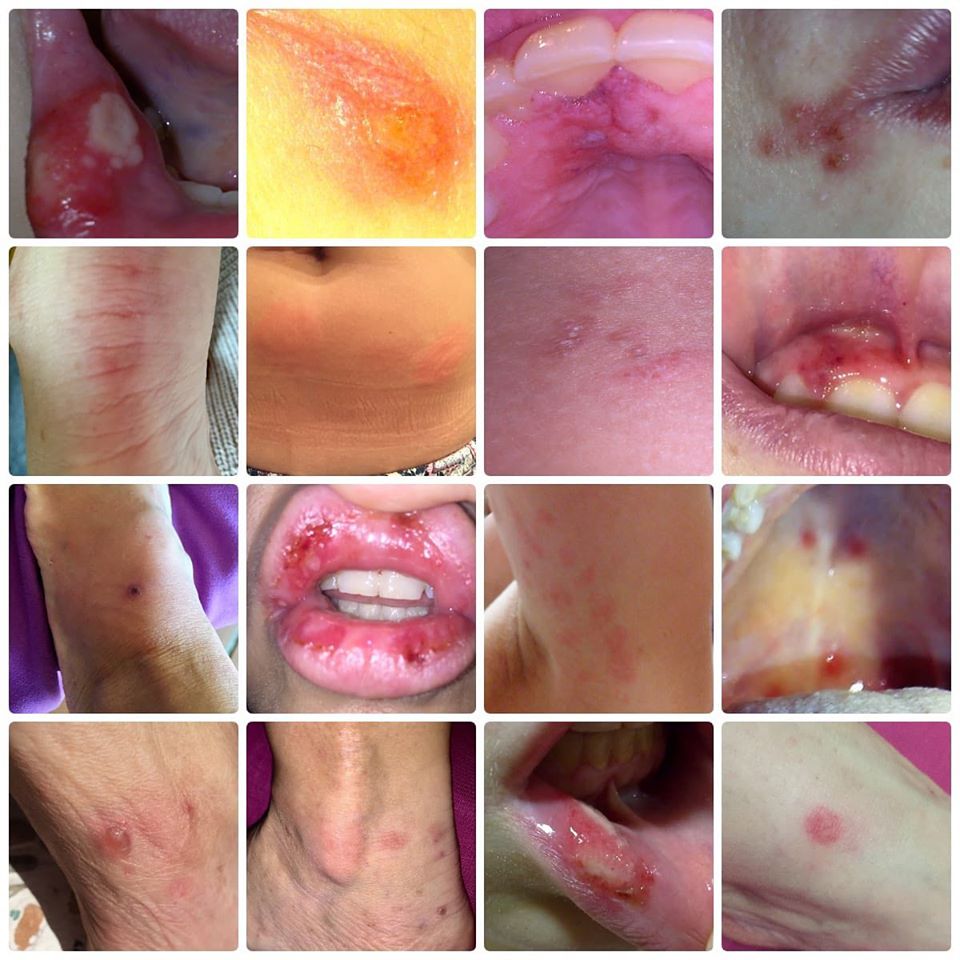
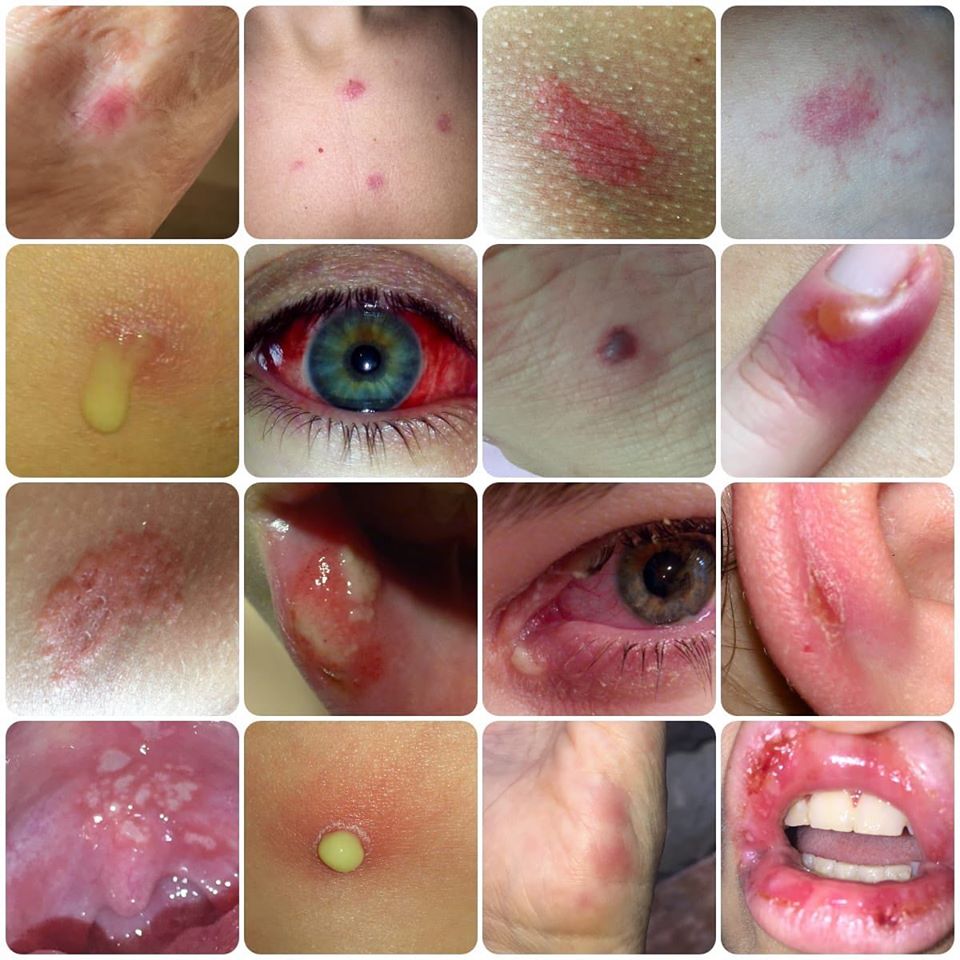
Además de esos síntomas, pueden ser comunes; adenopatías cervicales o los ganglios generalmente hinchados, dolor abdominal, vómitos, diarreas, cefaleas, migrañas basilares, vértigos periféricos, artritis de grandes articulaciones, erupciones en la piel y una minoría de pacientes (los que padecen HIDS más intensamente), divulga ulceras dolorosas, en la boca y en la vagina.
Los ataques son diferentes en cada persona. Otros síntomas que pueden aparecer durante un ataque son; agotamiento general del cuerpo, pérdida del apetito, dolor de músculos, dolores de cabeza, ulceras pequeñas en la boca y puntos rojos en la piel.
Después de estos ataques, el paciente se irá encontrando paulatinamente mejor, aunque le llevará varios días el volver a retomar su actividad y su energía.
Probablemente una crisis se unirá en algunas ocasiones con otra y esto puede llevar varias semanas de convalecencia para el paciente.
Algunos pacientes pueden tener graves episodios de fiebre, así como retraso de crecimiento y secuelas neurológicas.
Los ataques se repiten generalmente cada cuatro a seis semanas, pero el intervalo entre ellos puede variar sustancialmente de un paciente a otro.
La frecuencia de los brotes, es mayor durante la infancia y la adolescencia y van disminuyendo con la edad y aunque puede llegar a no presentarse durante años no llega a
desaparecer por completo. Con el paso del tiempo en ocasiones se reduce la intensidad de estos brotes, así como su frecuencia.
Los pacientes pueden estar libres de ataques por meses o incluso años. Pero no ha habido informes publicados, de que esto haya sucedido.
Los ataques empiezan de forma brusca y espontáneamente, habitualmente con escalofríos, palidez o incluso cianosis de los labios, dedos de las manos y pies, sensación de frio y convulsiones febriles.
¿Cuáles son los síntomas más habituales?
Fiebre que puede llegar a los 41º precedida de escalofríos y dolores de cabeza.
Poliartralgias, polimialgias y artritis que afectan a grandes articulaciones.
Adenopatías que se localizan en las cadenas ganglionares latero-cervicales unilateralea y dolorosas a la palpación.
Dolor abdominal. Habitualmente asociado a diarrea
Aftas orales y ulceras genitales.
Manifestaciones cutáneas como máculas eritematosas dolorosas, máculas-pápulas eritematosas no migratorias, eritema nodoso, urticaria o exantema morbiliforme.
Esplenomegalia: (aumento del bazo).
Complicaciones
Leves: adherencias abdominales y contracturas musculares
Graves: Amilodosis reactiva que se manifiesta en forma de disfunción renal desembocando en una insuficiencia renal crónica. Prevalencia de al menos el 10% de los pacientes
– Varios pacientes han llegado a decir que si cada vez que le ha dolido una parte del cuerpo se la hubieran extraído del mismo, no tendrían “organismo” –
¿Cuál es su tratamiento?
¿Tiene cura? El síndrome HIDS no tiene cura.
No tiene cura, ni dispone de ningún tratamiento efectivo que prevenga los ataques. Sin embargo, se están realizando investigaciones para encontrar una terapia efectiva y segura.
¿Cuánto tiempo dura la enfermedad? Este síndrome persiste toda la vida.
¿Cuál es el pronóstico de la enfermedad? Su pronóstico es bueno ya que la esperanza de vida no se ve reducida excepto en casos raros de infecciones graves o Amiloidosis renal.
Tiende a disminuir su gravedad con la edad en muchos pacientes.
¿Es contagiosa? Esta enfermedad no es contagiosa.
Es causado por un defecto en los genes que no pueden ser corregidos.
Lo único posible es controlar los síntomas de la fiebre y los dolores de los pacientes con medicamentos, como el paracetamol y otros pacientes aceptan efectos favorables de la prednisona (aunque no se recomienda que sea constantemente y en algunos casos puede aumentar la frecuencia de los episodios de la fiebre).
Testimonio de Mónica:«En mi caso, el paracetamol no ayuda demasiado cuando se tiene una fuerte crisis y después de haber probado durante 12 años consecutivos los corticoides, tuve que abandonar ese tratamiento porque deja bastantes efectos secundarios. Desde hace unos años he vuelto a usarlos, pero esta vez de manera esporádica y durante un periodo corto de tiempo y de esa manera me ayuda bastante a salir de las crisis».
Las personas que viven con esta enfermedad, sufren ataques de fiebre durante mucho tiempo, pero no visitan al médico cada vez que esto sucede, porque saben porque está ocurriendo esto y saben tratar su enfermedad. En caso de que un episodio de fiebre sea diferente del que es normalmente, si es más severo o con otros síntomas o que persista, entonces si hay que acudir al médico de cabecera o al especialista. Solo ellos podrán determinar si hay algo más.
La gente con HIDS por supuesto tiene tantas posibilidades de tener otras enfermedades como otra persona cualquiera.
No existe tratamiento para su curación pero si para paliar los síntomas de la enfermedad y por lo tanto, mejorar la calidad de vida de los pacientes.
Tratamientos, tales como
Simvastatim: en la mayoría de los pacientes disminuye el número de días de la enfermedad (aunque no paró todos los ataques de fiebre) ni su brusquedad. Es un medicamento usado a menudo por otras razones, tiene relativamente pocos efectos secundarios y no se observó ningún efecto secundario en los pacientes de HIDS.
Corticoides: administrando dosis elevadas al principio del brote provoca en un importante número de pacientes respuestas clínicas buenas o parciales, pero la administración prolongada puede dar lugar a indeseables efectos secundarios de mayor gravedad en pacientes pediátricos.
Anakinra: debe administrarse diariamente por vía subcutánea y su dosis puede ser nivelada al alza o a la baja en función de la intensidad de la enfermedad. Efectos secundarios: dolor y/o hinchazón en el punto de inyección seguido de un aumento de infecciones leves y moderadas del tracto respiratorio superior.
Rilonacept: se administra por vía subcutánea semanalmente. Efectos secundarios: los mismos que los del Anakinra, pero además desarrollo de anticuerpos dirigidos contra los fragmentos del receptor 1L-1R/IL-1 RacP de Rilonacept.
Canakinumab: se administra por vía subcutánea en intervalos de varias semanas. No se han identificado de momento desarrollo de anticuerpos anticanakinumab ni perdidas de eficacia del tratamiento por uso prolongado. Efectos secundarios: ligero aumento en la incidencia de infecciones habitualmente leves y localizadas en el tracto respiratorio superior y reacciones locales en el punto de inyección.
Utilizamos cookies para habilitar servicios y funciones esenciales en nuestro sitio, para recopilar datos de cómo interactúan nuestros visitantes con el sitio y para personalizar el contenido. Al hacer clic en "Aceptar las cookies" aceptas el uso de cookies en todas las páginas web. Al hacer clic en Rechazar o cerrar este banner, aceptas solo las cookies necesarias y no las cookies de analítica o de segmentación. Para obtener más información sobre nuestro uso de cookies, visita nuestra Política de Cookies. Puedes gestionar e informarte de tus preferencias de cookies en cualquier momento a través de la herramienta Configuración de Cookies de nuestro sitio. Puedes informarte más sobre las cookies que utilizamos aquí. AceptarRechazarAjustes
Politica de privacidad y cookies
Privacy Overview
This website uses cookies to improve your experience while you navigate through the website. Out of these, the cookies that are categorized as necessary are stored on your browser as they are essential for the working of basic functionalities of the website. We also use third-party cookies that help us analyze and understand how you use this website. These cookies will be stored in your browser only with your consent. You also have the option to opt-out of these cookies. But opting out of some of these cookies may affect your browsing experience.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.
Any cookies that may not be particularly necessary for the website to function and is used specifically to collect user personal data via analytics, ads, other embedded contents are termed as non-necessary cookies. It is mandatory to procure user consent prior to running these cookies on your website.