¿Qué es la Fiebre Mediterránea Familiar?
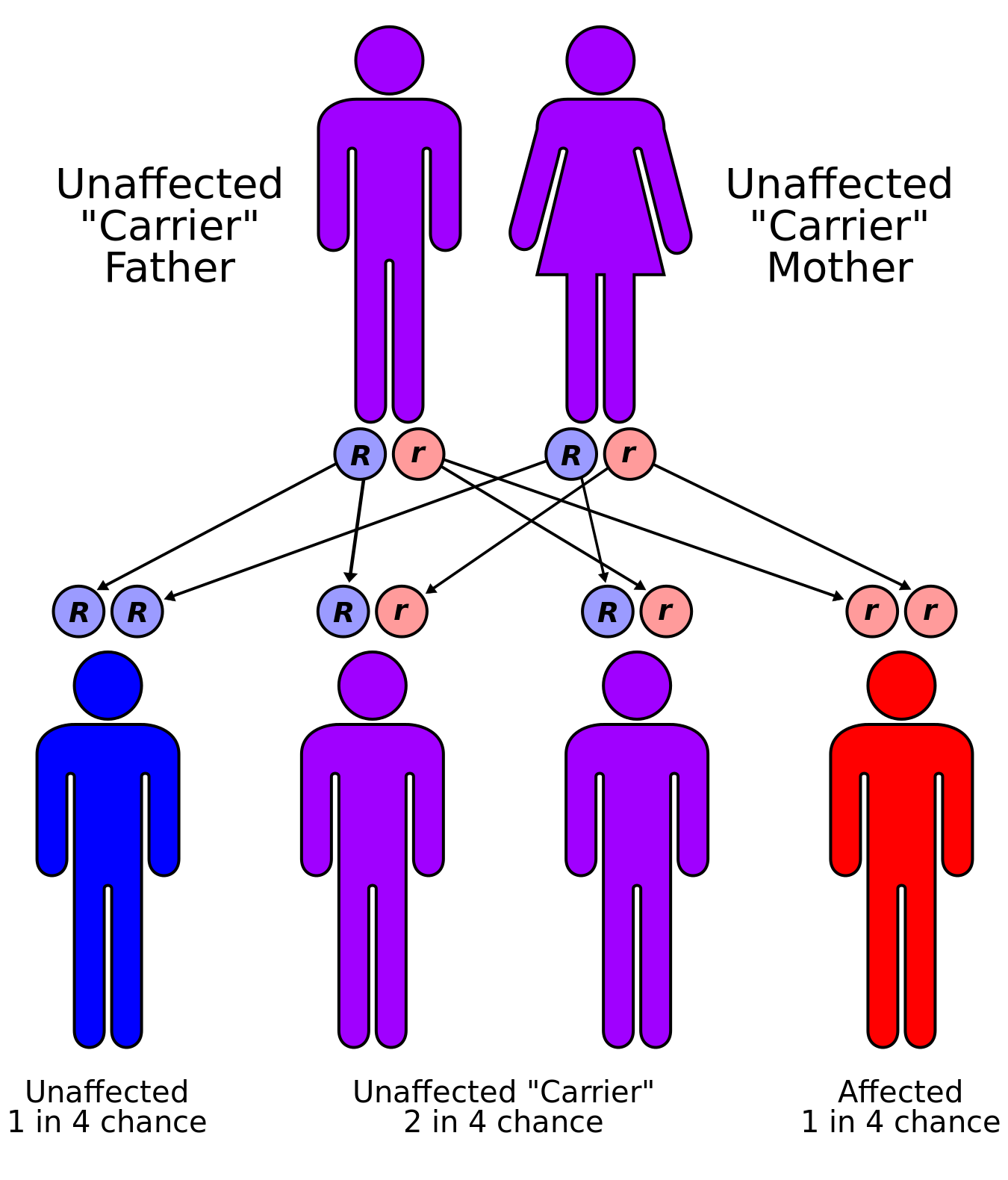
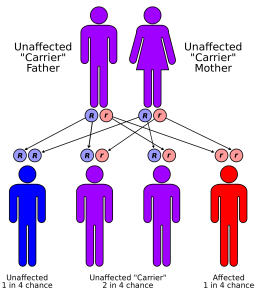
La Fiebre Mediterránea Familiar es una enfermedad genética autosómica recesiva y por tanto crónica. Esto significa que se hereda de padres a hijos y que para desarrollar la enfermedad es necesario recibir la copia materna y la copia paterna del gen mutado. No es contagiosa. Constituye el síndrome febril periódico más frecuente entre los síndromes autoinflamatorios y se encuentra dentro de las llamadas enfermedades raras ya que afecta a menos de 5/10000 habitantes en Europa.
Otros nombres para la Fiebre Mediterránea Familiar
La primera descripción de que se tiene constancia fue, posiblemente, la realizada en 1908 por Janeway y Mosenthal en una joven de 16 años con fiebre recurrente, dolor abdominal y leucocitosis. El alergólogo: Dr. Sheppard Siegal, describió por primera vez, en Nueva York, 5 casos con crisis de peritonitis en 1945. Como el curso de la enfermedad era esencialmente benigna la llamó: «peritonitis paroxística benigna». Mientras, el Dr. Hobart Reimann, que trabajaba en la Universidad Americana de Beirut, describe un cuadro más completo y lo llamó: “Enfermedad periódica”. En 1955 Heller introdujo por primera vez el término FMF debido a la frecuencia familiar y a su distribución geográfica. 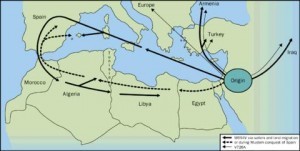
Esta enfermedad es, también, conocida como: Enfermedad Periódica, Fiebre periódica, Peritonitis Paroxística benigna, Poliserositis Paroxística Benigna, Síndrome Armenio, Poliserositis Paroxística Familiar, FMF, MEF, PPF, Síndrome Periódico Amiloide, Síndrome de Peritonitis Periódica, Poliserositis Recurrente, Enfermedad Periódica de Reimann, Síndrome de Reimann, Síndrome de Siegel-Cattan-Mamou, Trastorno Periódico, etc…
Los diversos nombres que se le han dado a la enfermedad han generado confusión respecto a su cuadro clínico. Hay que tener en cuenta que el nombre de: “Fiebre Periódica” también se utiliza para referirse a cualquiera de los Síndromes Autoinflamatorios de Fiebre Periódica. Ninguno de los nombres que se le han dado, incluido el de Fiebre Mediterránea Familiar es totalmente satisfactorio. Sin embargo, este último es el que habitualmente se utiliza con mayor frecuencia.
¿Cómo se produce una crisis?
El enfermo de FMF tiene alterado el mecanismo que regula la inflamación. El gen normal (MEFV) codifica una proteína, de 781 aminoácidos, que ayuda en la regulación de la respuesta inflamatoria al desactivar la respuesta inmune a nivel de la organización del citoesqueleto de leucocitos (el efecto terapéutico único de la colchicina en la FMF es dependiente de esta interacción) y que ha sido llamada de dos formas diferentes:
-
Pirina (nombre derivado de la palabra griega “pyros” utilizada para designar al fuego por su relación de este con la elevación de la temperatura provocada por la fiebre) por el Consorcio Internacional de FMF
-
Marenostrina (derivado de la palabra latina “Mare Nostrum” que se utilizaba para designar al mar Mediterráneo, lugar de origen de las poblaciones afectadas) por el Consorcio Francés de FMF.
En un enfermo de FMF el gen MEFV se encuentra mutado y no se fabrica esa proteína correctamente y por ello, se suceden episodios repetitivos de inflamación que suelen afectar a las serosas (membranas que recubren algunas cavidades como el tejido sinovial, la pleura o el peritoneo) de cualquiera de los órganos del cuerpo donde se encuentren y producir todos los síntomas posibles con relación a la inflamación de tales membranas. Se comportan como si estuvieran siendo agredidas y se produce una inflamación sin haber infección ni agresión por virus, bacterias, etc… Esta inflamación provoca los síntomas típicos incluyendo elevación de las “proteínas” de la inflamación en la sangre (Proteína C Reactiva o PCR) y del recuento leucocitario (leucocitosis).
No se ha aclarado completamente la función de la pirina, pero parece ser un supresor de la activación de caspasa 1, la enzima que estimula la producción de interleucina 1β, una citocina fundamental en el proceso de la inflamación. En otras palabras una pirina defectuosa no inhibe la inflamación normalmente,
No se sabe exactamente lo que desencadena los ataques, y por qué la sobreproducción de IL-1 llevaría a síntomas particulares en particular en ciertos órganos (por ejemplo, las articulaciones o la cavidad peritoneal).
Tipos de Fiebre Mediterránea Familiar
Lo característico de la enfermedad es que es cíclica. Se repite cada cierto número de días invariable si no hay tratamiento y en cada enfermo de forma característica. Generalmente no se presentan síntomas entre las crisis. La inflamación es de mayor o menor intensidad, pudiendo ir o no, acompañada de fiebre. Tanto es así que muchas veces la inflamación pasa inadvertida y otras veces los dolores son tan intensos que a muchos les han operado de apendicitis o de vesícula por los dolores abdominales tan fuertes que han tenido.
Existen tres tipos diferentes de FMF:
-
FMF tipo I: Es la enfermedad típica que cursa con síntomas: Cortos episodios recurrentes de inflamación y serositis, incluyendo fiebre, peritonitis, sinovitis, pleuritis y, en menor proporción, episodios de pericarditis, orquitis o meningitis.
-
FMF tipo II: Asíntomática. Sólo se descubre mediante un estudio genético positivo o cuando el enfermo ha desarrollado amiloidosis, la complicación más grave de la FMF, como primera manifestación clínica de la enfermedad en un individuo que no refiere otros síntomas.
-
FMF tipo III: Se refiere al estado de las personas heterocigotos “silenciosos” u homocigotos compuestos, en el que se detectan dos mutaciones en el gen MEFV sin signos o síntomas de la FMF ni de amiloidosis AA.
En los últimos años se ha observado que también los portadores de mutaciones heterocigotas pueden sufrir de una forma leve o incompleta de FMF, llamada “enfermedad similar a FMF”. La influencia de otros modificadores de genes y / o factores ambientales pueden contribuir a la penetrancia variable y a la variabilidad fenotípica de la FMF.
Diagnóstico de la Fiebre Mediterránea Familiar
Resulta indispensable diferenciar los cuadros clínicos de éstos enfermos de cuadros abdominales infecciosos, como apendicitis, pancreatitis u otros para no someterlos a tratamientos quirúrgicos inútiles. Además, como los síntomas son muy variados, muchos enfermos antes del diagnóstico son etiquetados de “enfermos psicosomáticos” y tienen que pasar por muchas consultas antes de ser diagnosticados. Es muy importante que el médico detecte a tiempo y sospeche esta enfermedad cuanto antes.
El diagnóstico de fiebre mediterránea familiar (FMF) se basa en varios factores:
- Manifestaciones clínicas típicas (síntomas y signos)
-
Respuesta positiva a la terapia de colchicina (remite la enfermedad con el tratamiento correcto)
-
Antecedentes familiares (es difícil cuando se carece de estos últimos o por la rareza de la enfermedad, el médico no tiene experiencia y no se puede diagnosticar lo que no se conoce)
-
Test de provocación de Metaraminol (MPT)
-
Pruebas genéticas.
Diagnóstico diferencial.
El diagnóstico diferencial se compone de los tres primeros factores: Manifestaciones clínicas típicas (síntomas y signos), respuesta positiva a la colchicina y antecedentes familiares.
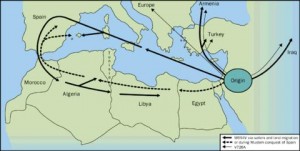
La consanguinidad en las familias de los afectados es un factor a tener en cuenta. Sin embargo, a veces no se conoce con exactitud, porque muchos enfermos provienen de grupos étnicos con gran endogamia. Afecta predominantemente a indivíduos con antepasados provinientes de los países de la cuenca mediterránea (España, Italia, Francia, etc…) de ahí su nombre. Hablamos de judíos (sefardíes más que askenazíes), armenios, árabes, etc… pero puede afectar a todo tipo de personas de todos los países debido a las migraciones producidas a lo largo de la historia. Se estima que una de cada 5-7 personas en los grupos étnicos afectados enumerados arriba pueden ser portadores del gen de la Fiebre Mediterránea Familiar, y una de cada 200 personas de estos grupos étnicos puede tener esta enfermedad.
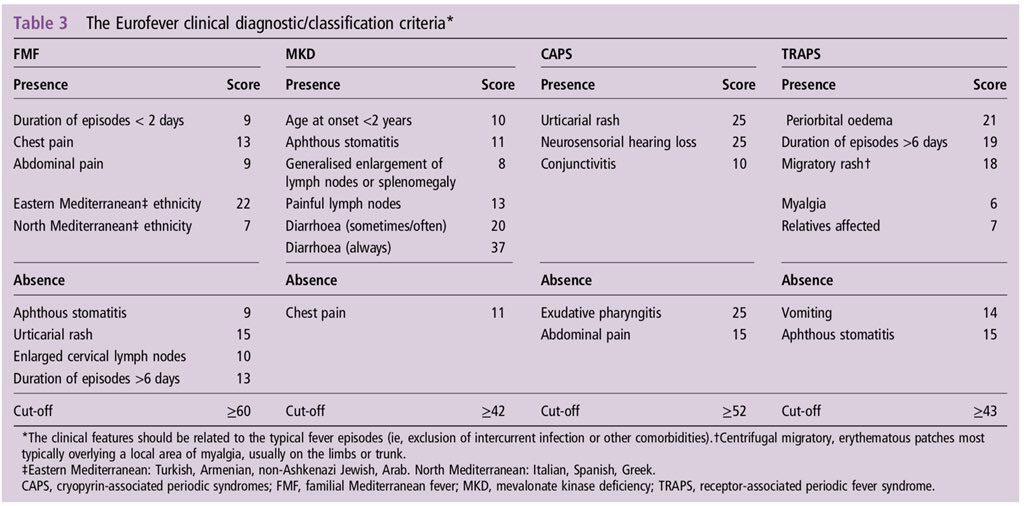
Para establecer un diagnóstico se establecen lo que se llaman: Criterios diagnósticos. Para la Fiebre Mediterránea Familiar existen diversos criterios aceptados internacionalmente. Un ejemplo serían los criterios de Tel-Hashomer, los criterios de Livneh y los criterios turcos.
En este enlace se puede leer un resumen de tales criterios: Criterios diagnósticos para la Fiebre Mediterránea Familiar


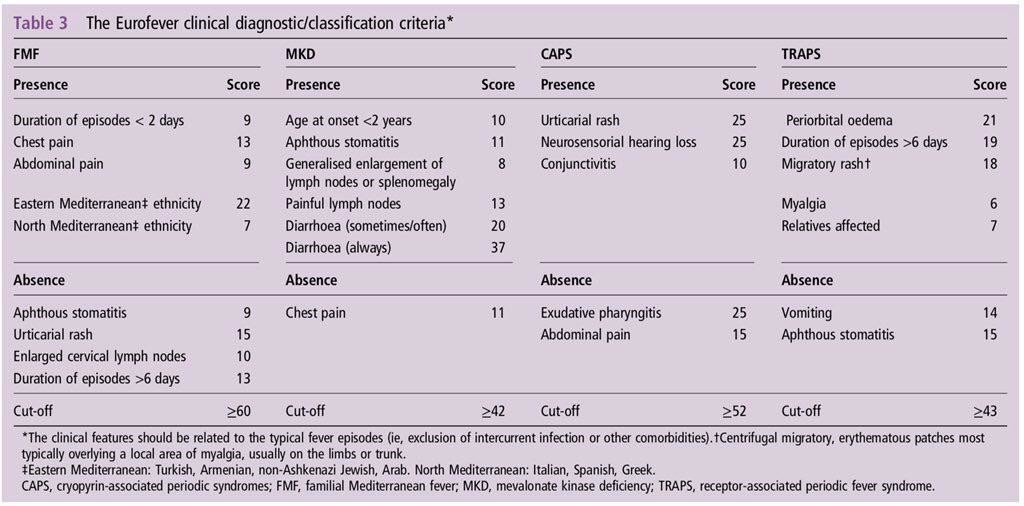 Criterios diagnósticos de EUROFEVER 2015
Criterios diagnósticos de EUROFEVER 2015
Ciertos análisis de sangre pueden arrojar resultados superiores a lo normal si se realizan durante una crisis. Estos pueden ser provechosos para diagnosticar, y supervisar el tratamiento en pacientes con el FMF y pueden incluir:
-
Analítica de proteína C reactiva
-
Tasa de sedimentación eritrocítica o velocidad de sedimentación globular (ESR)
-
Hemograma o conteo completo de células de la sangre (CBC o CSC)
-
Conteo de glóbulos blancos o leucocitos (WBC)
-
Análisis de fibrinógeno
-
Análisis del suero en sangre
Las analíticas o las radiografías pueden descartar otras posibles enfermedades para ayudar a determinar el diagnóstico.
Test de Provocación de Metaraminol (MPT)
Mención aparte merece el test de provocación de Metaraminol. Barakat, en 1984, publicó una prueba específica y altamente sensible de provocación diagnóstica. Consiste en la administración de 10 mg de una infusión de Metaraminol en suero fisiológico. Con esto se puede considerar un diagnóstico positivo de FMF si el enfermo presenta una crisis típica, aunque más leve. La crisis puede ocurrir entre 4 y 48 horas después de la administración y se da solamente en los individuos afectados. Sin embargo el test MPT es más específico que sensible, con lo que no identifica todos los casos de FMF aunque un MPT positivo puede ser muy útil.
No obstante el test no está exento de efectos secundarios cardíacos, y la elevación de la dopamina betahidroxilasa en el suero de los enfermos de FMF respecto a controles no ha sido confirmada.
Diagnóstico genético.
Sin embargo el diagnóstico diferencial resulta ser insuficiente y crear confusión en muchos casos ya que la Fiebre Mediterránea Familiar forma parte de los llamados Síndromes Autoinflamatorios. Este tipo de enfermedades son muy parecidas entre sí y es muy fácil errar en el diagnóstico.
En 1997, se identificó el gen causante de la FMF y se desarrolló una prueba genética mediante un análisis de sangre para estudiar las mutaciones. Su origen se encuentra en varias mutaciones genéticas del brazo corto del cromosoma 16. La mayoría de ellas cambian uno o varios de los 781 aminoácidos de la secuencia de la proteína mientras a unas pocas mutaciones les faltan pequeños fragmentos de ADN del gen MEFV que lleva a la síntesis de una pirina anormalmente pequeña. La mutación más común cambia el aminoácido metionina por el aminoácido valina en la posición 694 (escrito Met694Val ó M694V) en 16p13.3 del gen MEFV. Entre los enfermos de Fiebre mediterránes Familiar esta mutación está asociada con un incremento en el riesgo de desarrollar amiloidosis. Algunas evidencias sugieren que otro gen llamado SAA1 puede modificar el riesgo de desarrollar amiloidosis entre enfermos con la mutación M694V.
La Fiebre Mediterránea Familiar se hereda siguiendo un patrón recesivo
No obstante, en ocasiones, este análisis genético da negativo. Pese a la condición de recesiva de la Fiebre Mediterránea Familiar, se han descrito casos de personas con un sólo gen alterado que sí que padecen la enfermedad. Existen dos posibles explicaciones a este hecho:
-
Que el supuesto gen sano estuviera, también, mutado pero dicha mutación no se haya descubierto aún.
-
Que se trate de los pocos casos de Fiebre Mediterránea Familiar de herencia genética dominante que se han descrito. Los patrones de herencia son variables.
Lo más conveniente, es realizar esta analítica. Porque, aunque puede dar negativa al haber muchas variaciones genéticas aún no estudiadas, en caso de resultar positiva junto al diagnóstico diferencial, las posibilidades de padecer FMF son casi del 100%. Además es muy útil para:
El conocimiento de las mutaciones causantes, tiene importancia pronóstica y como guía del tratamiento ya que los portadores de determinadas mutaciones son los que tienen mayor riesgo de sufrir amiloidosis. De hecho se suele ofrecer la prueba genética molecular a todos los familiares de primer grado y otros miembros de la familia (independientemente de los síntomas) especialmente cuando la mutación M694V está presente debido al riesgo de amiloidosis renal que esta mutación conlleva. Por eso es muy importante hacer un estudio familiar ya que, al ser genética es hereditaria y se aplicaría a padres y hermanos principalmente. Para saber quien está enfermo y quien es portador y actuar en consecuencia.
En caso de estar planeando un embarazo hay que saber que si ambos padres son heterocigotos, el riesgo para los hijos de heredar dos variantes patógenas y ser afectado es del 25%. Se puede realizar una prueba de portador a los familiares de alto riesgo y una prueba prenatal para los embarazos con mayor riesgo si se conocen las variantes patógenas del gen MEFV en la familia.
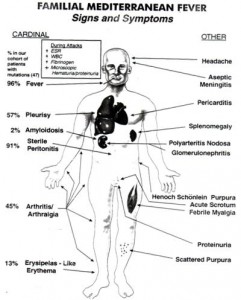
Síntomas de la Fiebre Mediterránea Familiar
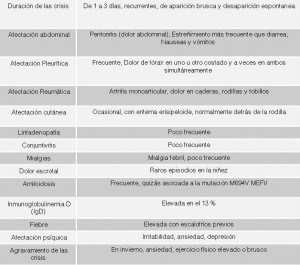
- Duración de las crisis: De 24 a 72 horas. Recurrentes. De aparición brusca y desaparición espontánea
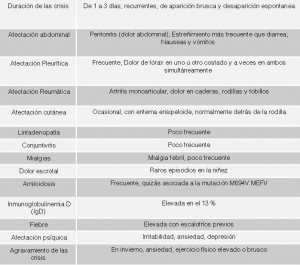
Tabla de síntomas (basada en un artículo del Dr. Daniel Kastner)
- Fiebre elevada que, por lo regular, alcanza su pico máximo entre las 12 y las 24 horas. A menudo con escalofríos previos. O episodios alternantes de fiebre y escalofríos. La mayoría de crisis cursan con fiebre pero no todas.
- Afectación abdominal: Dolor abdominal (peritonitis no infecciosa), estreñimiento (más frecuente que diarrea), nauseas y vómitos
-
Afectación Pleurítica: Frecuente. Dolor de tórax (pleuritis no infecciosa) en uno u otro costado y a veces en ambos simultaneamente que se intensifica con la inspiración.
-
Afectación Reumática: Dolor articular (artralgia) e inflamación articular (artritis generalmente monoarticular) por inflamación del tejido sinovial (sinovitis no infecciosa), Esto se traduce en dolor de caderas, rodillas y tobillos. Dolor muscular (mialgias) ocasional.
-
Afectación cutánea: Eritema erisipeloide (Úlceras o lesiones cutáneas rojas e inflamadas cuyo diámetro fluctúa entre 5 y 20 centímetros). Generalmente en la cara anterior de las extremidades inferiores. Sobre todo detrás de las rodillas.
-
Inflamación de las meninges: Meningitis no infecciosa. Es un síntoma poco frecuente
-
Inflamación del tejido conjuntivo del ojo (conjuntivitis) con dolor ocular. Es un síntoma poco frecuente
-
Afectación otorrinolaringológica: Inflamación no infecciosa de la faringe (faringitis) con dolor de garganta y aftas bucales, inflamación no infecciosa de los oídos (otitis), inflamación no infecciosa de los ganglios linfáticos (linfadenopatía). Son síntomas poco frecuentes
-
Afectación cardiaca: Inflamación no infecciosa del pericardio, tejido que recubre el corazón, (pericarditis) que se manifiesta con dolor en el pecho
-
Inflamación no infecciosa de los vasos sanguíneos (vasculitis y poliarteritis nodosa) con aparición de petequias y púrpuras. Púrpura de Schönlein-Henoch. Son síntomas poco frecuentes.
-
Dolor escrotal o testicular (orquitis), vaginal (vaginitis) o de ovarios. Se han descrito raros episodios en la niñez
-
Inmunoglobulinemia D (IgD): Elevada en el 13 % de los casos
-
Afectación psíquica: Irritabilidad, ansiedad, depresión, fatiga, alteración del sueño, inquietud, cambios de humor, fibromialgia.
Es interesante decir que los enfermos no tienen por qué experimentar todos los síntomas y que varían mucho de uno a otro e incluso, en el mismo individuo suele haber una gran variación (pueden experimentar tipos diferentes de crisis con mayor o menor gravedad) a lo largo de toda su vida.
La edad de aparición de estos síntomas es, en un 90 % de los casos en la preadolescencia y la adolescencia (alrededor de los 20 años) pero se han descrito muchos casos en la lactancia y la niñez.
Factores desencadenantes y agravamiento de las crisis
Existen ciertos factores que pueden desencadenar una crisis de FMF e incluso, empeorar la que ya se tiene. Estos factores son:
-
Estrés físico: El ejercicio físico intenso o brusco y los traumatismos (golpes y caídas)
-
Estrés psicológico: Ansiedad, falta de sueño
-
Las intervenciones quirúrgicas
-
Los partos. Se han descrito casos de mujeres con FMF asintomáticas hasta que han dado a luz.
-
Las enfermedades infecciosas. Probablemente porque altera la respuesta inmunológica del enfermo.
-
Menstruación: Normalmente es un periodo en el que se producen más crisis.
-
La alimentación: La grasas animales, el gluten (existen estudios que relacionan una dieta sin gluten con la mejora o remisión de los síntomas) y la lactosa (probablemente porque interfiere en la absorción de la colchicina aunque hay estudios que relacionan la toma de colchicina con una predisposición a desarrollar intolerancia a la lactosa).
-
Factores ambientales como el mal tiempo (estaciones frías) o temperaturas muy elevadas.
Logicamente, para evitar las crisis o un empeoramiento de las mismas, será necesario evitar este tipo de factores.
Tipos de crisis
Existen, básicamente, 7 tipos diferentes de crisis: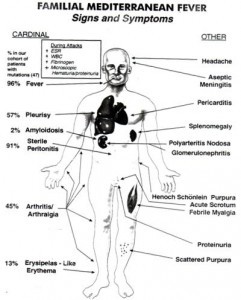
-
Crisis febriles aisladas de corta duración (entre 12 y 48 horas). Sin ningún otro síntoma. Se dan en el 25% de los enfermos.
-
Crisis abdominales expresadas por: fiebre, dolor abdominal agudo como de apendicitis. Afectan el abdomen entero con todos los signos de peritonitis (inflamación del revestimiento abdominal) y pueden conducir a innecesarias intervenciones de laparotomía. Estreñimiento o diarrea y náuseas. También se han descrito crisis incompletas con abdomen no tenso y análisis de sangre normales. Este tipo de crisis abdominales se dan en el 95% de los enfermos.
-
Artritis – Inflamación de las articulaciones caracterizada por: fiebre, hinchazón de las articulaciones y restricción de su función. Ocurren principalmente en las articulaciones grandes, Especialmente en las piernas. Por lo general, sólo una articulación se ve afectada. Este tipo de crisis se dan en el 75% de los enfermos
-
Crisis de pleuritis (inflamación de la pleura) manifestadas como dolores en un lado del pecho que pueden extenderse al otro lado o en ambos, intensificados con la inspiración (inhalación), respiraciones breves y fiebre. También pericarditis (inflamación del pericardio). Este tipo de crisis ocurre en el 30% o 40% de los enfermos.
-
Crisis dermatológicas como urticaria o erisipelas (dermatitis erisipeloide), que es una reacción de la piel en las piernas, son mucho menos comunes.
-
Crisis escrotales debidas a la inflamación de la Tunica vaginalis que pueden ser confundidas con escroto agudo (es decir torsión testicular) y ocurren en hasta un 5% de los enfermos.
-
Mialgia. Raro en aislamiento.
Registro de las crisis
Es muy recomendable llevar un registro de fechas con todos los detalles de las diferentes crisis del enfermo y con fotografías de los síntomas a ser posible, para poder enseñarlo al médico en cada visita. Este constituye una valiosa ayuda para el diagnóstico y el tratamiento de la Fiebre Mediterránea Familiar. Muchos enfermos llegan, de esta manera, a poder predecir la fecha de su próxima crisis.
Aquí puedes descargarte una Tabla de registro de crisis de FMF
Existen, también, aplicaciones para móvil que pueden ayudar a realizar este registro. Por ejemplo:
¿Existe un tratamiento efectivo para la Fiebre Mediterránea Familiar?
El tratamiento más habitual es la colchicina  que previene las crisis y la aparición de amiloidosis. El pronóstico con este medicamento es bueno ya que, como hemos comentado arriba, previene la amiloidosis (complicación grave de la enfermedad. Véase la sección de amiloidosis de esta misma página). Es muy seguro y tiene pocos efectos secundarios (diarrea,disminución de los glóbulos rojos, blancos, debilidad muscular, caída del cabello, pérdida de apetito) pero hay gente que no la tolera a la dosis óptima para controlar la enfermedad, desarrollando efectos secundarios y haciendo que la terapia con colchicina sea imposible. Otros no la toman y pueden desarrollar amiloidosis. Por eso es tan importante, la investigación de nuevos medicamentos para estas enfermedades raras (“medicamentos huérfanos”). Aún así existen sustitutos para la colchicina (Anakinra, Infliximab, Interferon alfa, Ilaris, etc…) Algunos enfermos experimentan sólo mejoría estos medicamentos alternativos lo que frena las crisis pero no la amiloidosis. Véase la sección de Tratamientos de esta web
que previene las crisis y la aparición de amiloidosis. El pronóstico con este medicamento es bueno ya que, como hemos comentado arriba, previene la amiloidosis (complicación grave de la enfermedad. Véase la sección de amiloidosis de esta misma página). Es muy seguro y tiene pocos efectos secundarios (diarrea,disminución de los glóbulos rojos, blancos, debilidad muscular, caída del cabello, pérdida de apetito) pero hay gente que no la tolera a la dosis óptima para controlar la enfermedad, desarrollando efectos secundarios y haciendo que la terapia con colchicina sea imposible. Otros no la toman y pueden desarrollar amiloidosis. Por eso es tan importante, la investigación de nuevos medicamentos para estas enfermedades raras (“medicamentos huérfanos”). Aún así existen sustitutos para la colchicina (Anakinra, Infliximab, Interferon alfa, Ilaris, etc…) Algunos enfermos experimentan sólo mejoría estos medicamentos alternativos lo que frena las crisis pero no la amiloidosis. Véase la sección de Tratamientos de esta web
¿Qué consecuencias tiene en la vida del enfermo?
El principal problema es llegar a un diagnóstico porque es una enfermedad difícil de diagnosticar y hasta que se llega a él, el niño/adulto, sufre mucho; de incomprensión, porque esta enfermo y sin el tratamiento, los brotes hacen perder jornadas de clase/trabajo, el enfermo ha de sufrir dolores y si no se diagnóstica y trata a tiempo, se desarrolla la amiloidosis.
Es importante concienciar al enfermo, una vez diagnosticado, de que tiene una enfermedad para toda la vida. Pero con el tratamiento correcto, que no debe abandonar nunca (la colchicina), con una buena alimentación (rica en frutas y verduras, pobre en grasas) y ejercicio físico adecuado, puede hacer una vida normal, con una supervivencia igual que el resto, puede estudiar, viajar, casarse, tener hijos, etc…
No se debe estigmatizar al enfermo, pero si hacerle comprender la necesidad de llevar un control médico y una medicación de por vida, cuidarse de no realizar esfuerzos físicos excesivos y de no exponerse al frío/calor excesivo, o a demasiado estrés emocional. La tranquilidad es muy importante. Puede necesitar ayuda psicológica y apoyo emocional en muchos momentos de su vida para sobrellevar mejor su enfermedad. Si quiere tener hijos, puede acudir al consejo genético, para que no lo trasmita a su descendencia.
(Basado en el artículo escrito en 2014 por Mónica Inmaculada Tortosa Fito, presidenta de la actual Asociación FMF ESPAÑA para la web: Raras pero no invisibles)
Véase la sección de artículos sobre FMF de esta misma página
Véase la sección de tratamientos para FMF de esta misma página
Aplicación android, en inglés, sobre FMF. Para teléfonos móviles
Artículo en Wikipedia sobre la Fiebre Mediterránea Familiar
Descárgate esta infografía sobre Fiebre Mediterránea Familiar